Pharmaceutical Methods
Publishing Quality Research & Reviews
Pharmaceutical Methods
Publishing Quality Research & Reviews
Review Article - (2023) Volume 14, Issue 1
Received: Feb 25, 2023, Manuscript No. PHMETHODS-23-94080; Editor assigned: Feb 28, 2023, Pre QC No. PHMETHODS-23-94080 (PQ); Reviewed: Mar 14, 2023, QC No. PHMETHODS-23-94080; Revised: Mar 16, 2023, Manuscript No. PHMETHODS-23-94080 (R); Published: Mar 21, 2023, DOI: 10.35248/2229-4708.23.14.242
The aim of this article is to assess and set all the items necessary to conduct properly a trial. Nowadays it is necessary to facilitate trial management by correct advices. The article aims to provide a guide for this. The example considers clinical trial conduction on Helicobacter pylori by its therapeutics assessment as an ease example of trial management and appropriate information to fulfil and guide any clinical trial conditions properly. Clinical trial management is the core part of the assessment on clinical trial. Advices on this seems to be meaningful to contact any clinical trial property. The article assesses property the step by step the part of a clinical trial. Helicobacter pylori is a day to day infection to fight its approach should be promptly revised.
matadorbet grandpashabet betist bahsegel klasbahis jasminbet hepsi bahis dinamobet betvole betpark betlike betboo sultanbet tulipbet padisahbet savoybetting goldenbahis maksibet fenomenbet jojobet tarafbet
Clinical Trial, Helicobacter pylori, Practitioner, Amoxicillin, Clarithromycin
During the clinical trial and before the data analysis, Standard Operation Procedures (SOPs), and Good Clinical Practice (GCP) must be guaranteed [1]. Nowadays helicobacter pylori is a common infection. Trial management could be useful to underline the efficacy and safety of this common health issues. Furthermore the article address to any prompt and efficient response in randomised controlled trial management. The example considers clinical trial conduction on Helicobacter pylori by its therapeutics assessment. In general, it outlines the items that are necessary to facilitate the clinical trial outlining and conduction. Its aim is to coordinate the clinical trial management in general using this useful example. For this reason the article has dual goals.
Investigational product assessment
It is a clinical trial on an investigational medical product. Its combination of medical products aims to prevent disease in human beings (aspirin ≤ 325 mg and antibiotics such as metronidazole 400 mg and clarithromycin 250 mg or amoxicillin 1 g and clarithromycin 500 mg and full-dose Protonic Pomp Inhibitor (PPI)) against ulcer bleeding in patients affected by H. pylori. (The aspirin increases the chance of ulcer bleeding). The effect of the medicine compares their curative effects against placebo; it verifies their pharmacological effects by comparing the number of bleeding events; it identifies the eradication of the infection in patients taking aspirin; the study doesn't consider metabolism. The study compares the efficacy of medicines against placebo. The study controls aspirin safety in the presence of H. pylori infection. The clinical trial is not a non-interventional clinical trial. It studies more than one medical product that has an authorisation in the member state concerned; all products prescribed are by their approval; all patients attend to a regular therapeutic strategy described in the protocol; the decision not to prescribe any inflammatory drug defect the inclusion of the patients; diagnostic and monitoring procedures like Computed Tomography study and discharge records monitor patients like in current practice, the breath test taken is out of routine clinical practice instead [2,3].
Protocol appropriateness
The appropriate clinical trial protocol template includes the 33 items deriving from SPIRIT 2013 Statement. It attends to Good Clinical Practice GCP roles. Furthermore, the protocol guidance and template for use in a Clinical Trial of an Investigational Medicinal Product (CTIMP) by the NHS Health Research Authority show the appropriateness of the protocol (Figure 1) [4].

Figure 1: Participants information sheet.
Nowadays aspirin (acetyl-salicylic acid) is widely used to protect against heart attack and stroke. A wide majority of the En- glish population aged over 60 uses this type of therapy. Even if its efficacy been studied for long, its long-term use is the cause of side effects like ulcer bleeding. It has killed 3000 patients during 2007 and it has been causing of 13000 hospital admission for the same reason. Additionally, Helicobacter pylori (H. pylori) are the reason for the most common infection that attacks stomach and duodenum. Retrieved clinical data suggests that low dose aspirin increases the chance to cause ulcer bleeding in patients infected by H. pylori [5].
Trail features
Its treatment gives an effective eradication. It consists in “7-day twice-daily course of treatment consisting of a full-dose PPI, with either metronidazole 400 mg and clarithromycin 250 mg or amoxicillin 1 g and clarithromycin 500 mg and full-dose Protonic Pump Inhibitors (PPI) as per the National Institute for Health and Clinical Excellence (NICE) directives foresees.” The aim of this research ensures the evaluation of the efficacy of anti-ulcer bleeding antibiotic therapy due to H. Pylori during low dose aspirin co-assumption with a full dose of Protonic Pump Inhibitors (PPI). A further confirmation of the eradication derives by Computed Topography (CT) scan study. All adults aged 60+ take aspirin regularly and results positive to a breath test can participate in the survey. The participation to research is entirely voluntary [6].
It is a post-marketing analysis on the efficacy of antibiotic and full-dose Protonic Pomp Inhibitors (PPI) during low dose aspirin assumption against placebo (no therapeutic effect). The double-blind procedure does not allow recognising whether the medicine administered is placebo or not by part, the patients or health care professional. Patients will be divided into two equal groups, building the randomization (division). A secure web system tool guarantees an appropriate randomisation process. Any suspect adverse vent allows the unbinding procedure which is the unmasking procedure. During the interview, patients will reply if they are available to participate to the study signing the informed consent. Research nurse schedules them an appointment to their local General Practitioner (GP). During clinical trial execution, patients need to contact hospital promptly. They will be able to execute a Computed Topography (CT) scan to confirm the diagnosis within 24 hours. During clinical trial conduction a member of the study team assesses the patients. They receive informed consent and after having been confirmed their eligibility. The research team collected baseline data after the administration of the breath test. By the data regulation policy, guaranteeing confidentiality, the central laboratory in Liverpool executes samples' analysis, sending the results to the research team and local General Practitioner (GP). Investigational Medical Product (IMP) is e-held at local General Practitioner (GP) during the distribution period at 4°C as also at patients' home for quality proposes. Due to the double-blind procedure, medication packaging is the same in both arms of the trial. Patients become allocated to a medication ID number during the randomisation process. The patients receive the medication carrying their corresponding ID [7].
Patients receive the medicine within two weeks from the breath test. They receive the dose for one week. Research team contact patients after one week by phone, mail or how they have preferred to contact. After 12 months of the one-week treatment period, follow up data will be collected for all patients from their medical General Practitioner (GP) records.
Further, participants have the right to refuse to continue the clinical trial. Results dissemination can be used to obtain a public service improvement [8].
Informed consent gastric ulcers bleed reduction in patients taking aspirin
All tasks are sequential, so they depend one from another. The documents submission period for ethic approval, CTA approval, and R&D, is fundamental to obtain the permission and the final approval. Recruiting starts at the end of the 9th month (Figure 2 and Table 1) [9].
| SET UP TASKS | WEEK 1 | WEEK 2 | WEEK 3 | WEEK 4 | WEEK 5 | WEEK 6 | WEEK 7 | WEEK 8 | WEEK 9 |
|---|---|---|---|---|---|---|---|---|---|
| Trial planning and design |
|
||||||||
| Risk assessment |  |
||||||||
| Sponsorship |  |
||||||||
| Protocol development |  |
||||||||
| Feasibility and investigator selection |  |
||||||||
| Confirm sponsor |  |
||||||||
| Trial master file and eudract number assignation |  |
||||||||
| CTA submission |  |
||||||||
| Ethic submission |  |
||||||||
| R&D submission |  |
||||||||
| Permission and approval obtained |  |
||||||||
| Recruitment |  |
||||||||
| Treatent |  |
||||||||
| Follow-up |  |
||||||||
| Publishing |  |
Note: ( ) Trial planning and design, () Risk assessment, () Sponsorship, () Confirm sponsor, () Trial master file and eudract number assignation, () CTA submission, () Ethic submission, () R&D submission, () Permission and approval obtained, () Recruitment, () Treatent, () Follow-up, () Publishing
) Trial planning and design, () Risk assessment, () Sponsorship, () Confirm sponsor, () Trial master file and eudract number assignation, () CTA submission, () Ethic submission, () R&D submission, () Permission and approval obtained, () Recruitment, () Treatent, () Follow-up, () Publishing
Table 1: The essential timeline schedule.

Figure 2: Consent form for Gastric ulcers bleed Reduction in Patients taking Aspirin. Note: It is set by Gantt chart: The schedule assesses an example of a timetable.
The Sponsor is the organization, or the government body pays and oversees the economic approval. It provides the necessary funding to conduct the clinical trial. The clinical trial is object of its support by of the financial condition to conduct it. The clinical trial economic feasibility, analysis during the set-up period, aims to reduce the costs.
The Research Ethics Committee (REC) is responsible of the clinical trial ethical approval. It aims to conduct the clinical trial on a human subject in ethic manner, to safeguard the right well-being, safety, and dignity of participants by the convention of Helsinki. Its participation to trial is to fix the ethics principles could involve patients during participation. It evaluates participant eligibility as well. Research Ethic Committee REC must review ethic items of the research protocol, submitted by the study team, to guarantee ethical standards in trial conduction. It analyses the participants considering their characteristic required by the clinical trial regulations [10].
The Health Research Authority HRA assigns the approval of a clinical trial on the EC Clinical Trial Directive (2001/20/EC). As this research project is a clinical trial of an investigational medical product (as demonstrated yet in question)
• It needs to receive the approval by Health Research Authority HRA. Further, the Sponsor itself submits the protocol for clinical trial approval.
• Project filter accurately select the data to represent the project to be inserted in the data collection to assure quality and appropriateness.
• Questions in the dataset are sure that form completed correctly.
• Supporting documents make sure that all the documents supporting the clinical trial automatically added to the checklist.
• Prepare a checklist adding a full list of necessary materials and add the additional ones at the bottom.
• Make sure that the file has a correct version, date, file number.
• Make sure that the validation is possible.
• Submitting your application is sure it is possible to do it correctly and electronically.
• Submission amendments, schedule submission amendments if necessary.
• Form submitted to ensure that resolution queries are possible after the application for the approval. It is the regulatory authority assigns Clinical Trial Authorization CTA as it is in the Gantt chart.
Furthermore, this trial needs the R&D submission and the approval from the local NHS R&D office as NHS patients and NHS staffs take part to it.
Medicine and Healthcare Products Regulatory Agency MHRA approve Clinical Trial of an Investigational Medicinal Product (CTIMP) like in this case. It is registered on the European Clinical Trials Database by Clinical trial submissions can be made through Common European Submission Portal (CESP) obtaining a EudraCT number. (The EU Clinical trial website makes the information available online). Medicine and Healthcare Products Regulatory Agency MHRA approves trails like local governate authority through Integrated Research Application System IRAS. The application can be modified and withdrawn before the final assessment. The application must contain notification scheme, fees, and contact details. Additionally, an expert can assist the application.
Risks likelihood impact
Hospitalization for ulcer bleeding could be a suspected adverse event: The likelihood of ulcer bleeding could be higher in the placebo group rather than in the antibiotic group, because placebo have no effect. Breath test would confirm the prognosis and Computed Topography (CT) scan would rate the severity of the event. This procedure gives a chance to advise the research team to unbind the trial on time because of any suspected adverse event solution. It impacts on participants’ comfort. In case of the suspected adverse event, data should report participant’s health status as foreseen for ethic reason and break blinding procedure for risk solution.
An unbinding procedure due to suspected adverse event is the reason of withdrawal/discontinuation and data integrity: The statistical measure can assess ulcer bleeding. In case of withdrawal, the statistician can provide a variable analysis to determine the integrity of data due to patients’ discontinuation.
The loss of participants can affect the statistic value of the trial without any appropriate statistical arrangements; it is not possible to provide an assurance of quality during data collection.
Consequently, loss of follow up is a second issue for data integrity: Follow up loss must be calculated on any patients at time to adopt any improvement if they abandon the clinical trial. An amendment to the standard procedure can prevent data damage to set up modification. It must be foreseen by the protocol and approved by the competent authority. Internal incident reporting form must be submitted to the regulatory authority as well. It evaluates risks. It reviews the trial organization to plan the reaction and report it.
The loss of follow up on a patient could modify data value. It is due to his/her withdrawn/discontinuation. Statistical adjustment after amendment review is the best way to consider the impact on data analysis. It provides an improvement to set up finding the data collection process.
Standard operation procedure
Standard Operation Procedure (SOPs) assures quality in clinical trials. Good Manufacturing Practice (GMP), research team can recognize breath test batch number for the patient has taken it. As the batch number must be reported on the box. All patients corresponding to the batch number must be recalled. They can set up a new appointment to repeat the test. Amendments set up again trial conduction. The incident report form evaluates risk, review the trial organization, plan the reaction, and report the action planned to face the emergency. The Chief Investigator CI is the main figure involved in the decision- making process. Due to the internal incident reporting the Chief investigator CI can communicate to
• the Sponsor,
• the Research Ethic Committee REC,
• Health Research Authority HRA, and to the local R&D within the NHS the substantial amendment due to breath tests issues.
Further, the research team should a report of the impact of this issue during recruiting. Additionally, the Data Monitoring Committee must be advised of any procedure variation can differ the planned original data collections. At end, the whole research team is made aware of all the difference. Afterwards, it is also made aware of the procedure change adoption [11,12].
Randomization process management
Patients are allocated by a medication ID during the randomisation process. The patients receive medication by their ID number corresponding to the ones reported on the medicine boxes. The boxes containing placebo and antibiotic are not distinguishable at all. These procedure guarantees the anonymity of the treatment in a double-blind process by either patient side either research nurse or General Practitioners (GP). After twoweek breath test confirmed the diagnosis, the General Practitioners (GP) handle around the boxes to the patients during the treatment. Moreover, the patient will be asked whether they want to be contacted by phone, mail or test keeping; their number assigned during the visit. The double-blind procedure guarantees observation bias minimisation ensuring data quality collection, for the quality assurance proposes. The protocol must report the whole process in the blinding/masking section. The double-blind randomized controlled procedure should be declared in the protocol statement. All the described methods stand in the protocol to obtain the approval by and by the competent regulatory authorities.
This article aims to consider the characteristic of trail management. Suggestions given can be applied to any clinical trial. They aim to lead through a trail appropriate conduction. Although, the study team should provide support against study discontinuation, patient can retire at any time. Research nurses, as part of the research team, can review data anticipating the analysis discussion to rescue participants from severe health condition. It guarantees Standard Operation Procedures (SOPs), and Good Clinical Practice (GCP) during clinical trial participation or even due any suspected adverse event by the subsequent twelve months of the one-week treatment period. In conclusion, these two procedures allow the access to clinical trial data in advance.
[Google scholar], [Crossref], [Indexed].
[Google scholar], [Crossref].
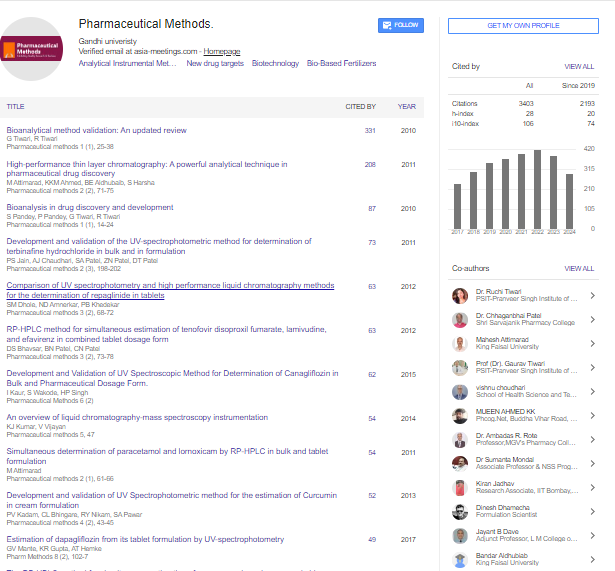
Pharmaceutical Methods received 3403 citations as per google scholar report