Pharmaceutical Methods
Publishing Quality Research & Reviews
Pharmaceutical Methods
Publishing Quality Research & Reviews
Short Communication - (2024) Volume 15, Issue 1
Received: Feb 21, 2024, Manuscript No. PHMETHODS-24-132178; Editor assigned: Feb 23, 2024, Pre QC No. PHMETHODS-24-132178 (PQ); Reviewed: Mar 08, 2024, QC No. PHMETHODS-24-132178; Revised: Mar 15, 2024, Manuscript No. PHMETHODS-24-132178 (R); Published: Mar 22, 2024, DOI: 10.35248/2229-4708.24.15.263
In the branch of pharmaceutical formulation, the integration of human plasma presents unique challenges and opportunities. Human plasma, the liquid component of blood, is a complex matrix containing a myriad of proteins, lipids, hormones, and other bioactive molecules. Understanding the interactions between pharmaceutical compounds and plasma components is essential for optimizing drug delivery systems, ensuring therapeutic efficacy, and minimizing adverse effects. In this article, we delve into the complications of pharmaceutical formulation in the context of human plasma and explore innovative strategies to enhance drug performance [1].
Human plasma serves as a significant medium for drug transport and distribution in the body. Upon administration, pharmaceutical compounds encounter various plasma proteins such as albumin, globulins, and fibrinogen, which can influence their pharmacokinetic behavior. Protein binding, or the reversible association of drugs with plasma proteins, plays a significant role in determining drug distribution, metabolism, and elimination. High protein binding can lead to reduced drug bioavailability and increased susceptibility to drug-drug interactions. Therefore, understanding the extent and nature of protein binding is essential for predicting drug behavior in vivo. One of the key challenges in pharmaceutical formulation involving human plasma is achieving optimal drug-protein interactions. While protein binding can enhance drug stability and prolong circulation time, excessive binding may limit drug diffusion into target tissues and impede therapeutic efficacy. Thus, having balance between protein binding and free drug fraction is critical for achieving desired pharmacological outcomes. Formulation strategies such as nanoparticle encapsulation, prodrug design, and molecular modification can be employed to modulate drug-protein interactions and enhance drug delivery efficiency. Nanotechnology has emerged as a promising approach for formulating drugs in the presence of human plasma. Nanoparticles, ranging in size from 1 to 1000 nanometers, offer unique advantages such as high surface area-to-volume ratio, tunable surface properties, and the ability to encapsulate both hydrophilic and hydrophobic drugs. By encapsulating drugs within nanoparticles, researchers can protect them from enzymatic degradation, prolong their circulation time, and improve their bioavailability.
Furthermore, surface modification of nanoparticles with biocompatible polymers or targeting ligands can facilitate selective drug delivery to specific tissues or cells, minimizing off-target effects and enhancing therapeutic efficacy. Prodrug design is another strategy employed in pharmaceutical formulation to optimize drug-plasma interactions. Prodrugs are bioreversible derivatives of drugs that undergo enzymatic or chemical transformation in vivo to release the active parent compound. By conjugating drugs with functional groups that enhance plasma stability or promote protein binding, researchers can modulate drug pharmacokinetics and improve therapeutic outcomes. Prodrugs can also be designed to target specific plasma proteins, thereby facilitating site-specific drug delivery and minimizing systemic side effects [2-5].
In addition to nanoparticle encapsulation and prodrug design, molecular modification of drug molecules offers a versatile approach to enhance drug-plasma interactions. Structural modifications such as the addition of hydrophilic or lipophilic moieties, alteration of stereochemistry, or introduction of functional groups can influence drug solubility, protein binding affinity, and metabolic stability. Rational drug design guided by computational modeling, structure-activity relationship studies, and in vitro screening assays enables researchers to tailor drug molecules for optimal performance in the presence of human plasma. Furthermore, advances in analytical techniques such as mass spectrometry, nuclear magnetic resonance spectroscopy, and surface plasmon resonance have facilitated the characterization of drugprotein interactions at the molecular level. These techniques allow researchers to elucidate binding affinities, binding sites, and kinetic parameters of drug-plasma protein interactions, providing valuable insights for rational drug design and formulation optimization. Moreover, high-throughput screening platforms and computational modeling approaches enable rapid screening of drug candidates and prediction of their behavior in complex biological systems. Understanding the complex interactions between pharmaceutical compounds and plasma proteins is essential for optimizing drug delivery systems and enhancing therapeutic efficacy. By employing innovative formulation strategies such as nanoparticle encapsulation, prodrug design, and molecular modification, researchers can modulate drug-plasma interactions and improve drug performance in vivo. Furthermore, advances in analytical techniques enable comprehensive characterization of drug-protein interactions, facilitating rational drug design and formulation optimization.
[Crossref] [Google Scholar] [Indexed]
[Crossref] [Google Scholar] [Indexed]
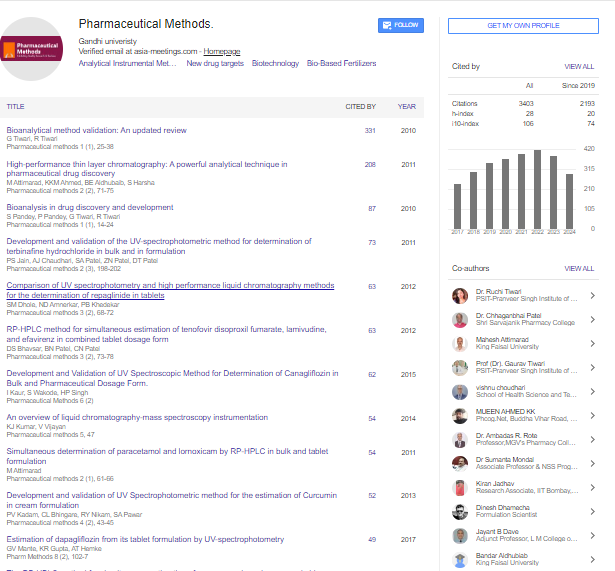
Pharmaceutical Methods received 3403 citations as per google scholar report